
Using the beta-lactamase TEM-1 involved in the spread of antibiotic resistance as a model, we explored, in a work published in august 2013 in PNAS, the mutational landscape of an enzyme by producing and sequencing a collection of 10,000 mutants. We established that mutation type, solvent accessibility of residues, and the predicted effect of mutations on protein stability primarily determined alone or in combination changes in minimum inhibitory concentration of mutants. This work thereby provides an integrated framework to study mutation effects and a tool to understand/define better the epistatic interactions.
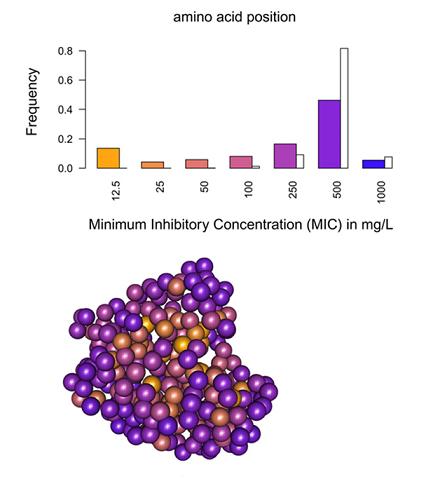
A figure showing the distribution of mutation effects on the MIC of ampicillin and the average effect for each residue on the 3D structure of the protein
In collaboration with the team of Antoine Andremont, we published in the february 2013 issue of Environmental Microbiology Reports a large-scale epidemiological survey of commensal Escherichia coli in Trois-Sauts, an isolated village located in the south of French Guiana where human population exchanges are restricted and source of antibiotics controlled. A clear genetic structure between E. coli populations was observed with human strains belonging very rarely to B2 phylogroup, exhibiting few virulence genes and bacteriocins but being antibiotic resistant whereas wild animal strains were characterized by 46.1% of B2 phylogroup belonging, with very unique and infrequent sequence types, numerous extraintestinal genes and bacteriocins but no antibiotic resistance; the human-associated animal strains being intermediate. The existence of such E. coli structured phylogenetic diversity within various hosts of a single localization has never been reported.

Photo of the site of sampling in the Trois-Sauts region, French Guiana, with the transects in the pristine forest in red.
In a collaborative work with Brandon Gaut and Tony Long team at the University of California Irvine, published in january 2012 in Science, we have caught natural selection in the act. We experimentally evolved 115 populations of a strain of Escherichia coli for 2000 generations under extreme conditions, to 42.2°C, and then completely sequenced genomes of these 115 lines in search of mutations. We identified 1331 total mutations, affecting more than 600 different sites on the genome. Few mutations have been found shared between the lines, but a strong pattern of convergence emerged at the level of genes, operons and functional complexes. Indeed, mutations were found at different nucleotides in the same genes, the same sets of cotranscribed genes or in genes coding for proteins that interact with each other in the same functional pathway. This is a strong evidence that these mutations were selected during evolution in the laboratory and allowed lines to adapt. It is interesting to note that the mutations were identified in global regulators such as RNA polymerase or transcription termination factor Rho. Finally, we showed that these mutations had preferential associations, indicating the presence of epistasis, i.e. interactions between mutations. Thus, adaptation occurred through many different genetic paths, but by affecting the same functions.
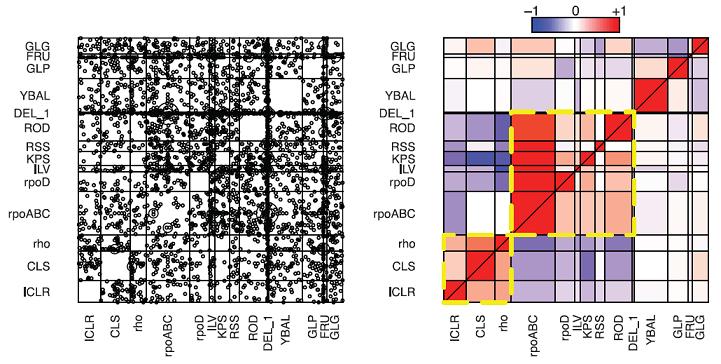
A figure showing the non-random occurrence of the mutations in the various operational units
Our laboratory has conduced a French multicentric prospective study on more than 1,000 E. coli bacteraemias during 2005, the COLIBAFI study. We have recently published 2 papers on the adult patients in 2011, one in the Journal of Clinical Microbiology and the other in Clinical Microbiology and Infection. We showed in the first paper that E. coli bacteraemia is still a severe disease, with 13% of mortality. Multivariate analysis indicated that host factors and portal of entry outweigh bacterial determinants to predict the severity of the bacteraemia. In the second paper, we analysed the prevalence, molecular epidemiology and clinical features of the bacteraemias caused by third-generation cephalosporin-resistant E. coli. 3.8% of the strains have a decreased susceptibility to third-generation cephalosporins, half by extended-spectrum beta-lactamases and half by AmpC and OXA-type penicillinases. Underlying disease and prior use of antibiotics were independent risk factors for development of a third-generation cephalosporin resistant strain bacteraemia. Infections with third-generation cephalosporin resistant strains, as compared with those caused by their third-generation cephalosporin sensitive counterparts, were more severe.
In two multi-team collaboration works published in 2011 in the Journal of Bacteriology and the Journal of Evolutionary Biology, we have linked the genomic elements to phenotypic behaviours in natural isolates of E. coli. We have first developed a tailor-made reconstruction strategy to build metabolic networks from whole genome sequences. We found that among the 1,545 reactions forming the E. coli pan-metabolism, 57% are conserved among all the strains (core-metabolism), in sharp contrast with the core-genome (see below PLoS Genetics 2009). Core reactions were significantly overrepresented among biosynthetic reactions compared to the more variable degradation processes. We have then studied the growth yield on carbon sources of the strains. The presence/absence of metabolic pathways, which was linked to the phylogeny, explained most of the growth capacities. However, few discrepancies, possibly caused by gene regulatory effects, blurred this link and produced a continuous phenotypic diversity.
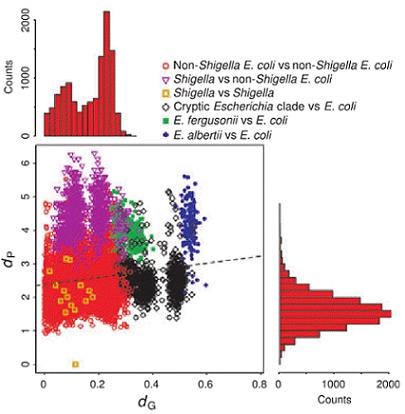
Relationship between the metabolic distance, dp, and the genetic distance, dg, resulting from comparisons between 159 E. coli strains (comprising 6 Shigella strains), 6 cryptic Escherichia clade strains, 2 E. fergusonii strains and 1 E. albertii strain. The histograms represent the distribution of dg (top) and dp (right) for the comparisons between 2 non-Shigella E. coli strains corresponding to the red circles on the plot.
In the september 2010 issue of PLoS Pathogens, we investigated whether an infection is a site of pathogen within-species diversity. Our results indicate that there is indeed extensive diversity during human extraintestinal infections by Escherichia coli. This diversity was of two types, not mutually exclusive, as we found that patients were infected either by several distinct E. coli clones or by members of a single clone that exhibit micro-heterogeneity. The high degree of phenotypic diversity, including antibiotic resistance, suggests that there is no uniform selection pressure leading to a single fitter clone during an infection. We discuss a possible mechanism and a mathematical model that explains these unexpected results, based on the trade off between self-preservation and nutritional competence (SPANC) and different levels of RpoS. Our data suggest that the evolution of diversity in the course of an infection and in in vitro experimental evolution in the absence of host immune selective pressure may have many parallels. Whatever the mechanisms leading to diversity, our results have strong medical implications in terms of the need for more extensive isolate testing before deciding on antibiotic therapies.
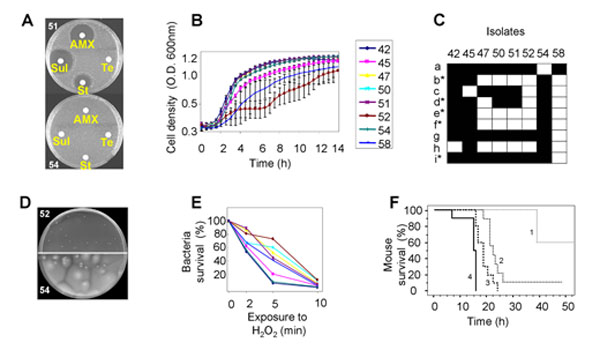
Illustration of the phenotypic diversity observed in the E. coli isolates of a patient with liver abscess infected by a unique clone.
(A) Variability in the antibiotypes with a plasmid loss in isolate 51 responsible for the susceptible phenotype. AMX: amoxicillin, Sul: sulfonamide, St: streptomycin, Te: tetracycline. (B) Growth curves in Luria Bertani broth at 37°C of 8 isolates, showing impaired growth of isolates 45, 52 and 58. (C) Carbon source utilisation using Biolog GN2 plates that test 95 substrates. Only substrates differentially used by the isolates are indicated. The substrates are as follows. a: Dextrin, b: Bromosuccinic Acid, c: Glucuronamide, d: L-Alanine, e: L-Asparagine, f: L-Aspartic Acid, g: Glycyl-L-Aspartic Acid, h: L-Serine, i: D,L-α-Glycerol Phosphate. Substrates with a star correspond to substrates whose metabolism is stimulated by an rpoS disruption. Black and white squares indicate the use or the absence of use of the substrate, respectively. (D) Non motile (52) and motile (54) isolates in 0.35% agar plates incubated for 48 hours at 37°C in humid atmosphere. (E) Sensitivity to H2O2 measured by the survival in % at different times. The colour code for the isolates is as in (B). (F) Kaplan-Meier curves estimating the survival function of mice subcutaneously inoculated by 2 X 108 colony forming units of the different isolates. Only one curve of each statistically significant category is presented.
In a collaboration with Médéric Diard and Ivan Matic from the Tamara lab (http://www.necker.fr/tamara/) we have investigated in two 2010 papers, one in the International Journal of Medical Microbiology and the other in the Journal of Bacteriology the role of the E. coli pathogenicity islands (PAIs) by testing the effects of simple and multiple deletion mutants of the uropathogenic E. coli 536 strain in various models. We showed that PAIs cooperate in an additive manner to achieve extra-intestinal virulence but also are fitness elements involved in the intestinal colonization, demonstrating experimentally the coincidental evolution hypothesis postulating that extra-intestinal E. coli virulence is a by-product of commensalism. This effect of PAIs on intestinal commensalism seems to be linked to the hypermotility observed for the mutant devoid of all PAIs.
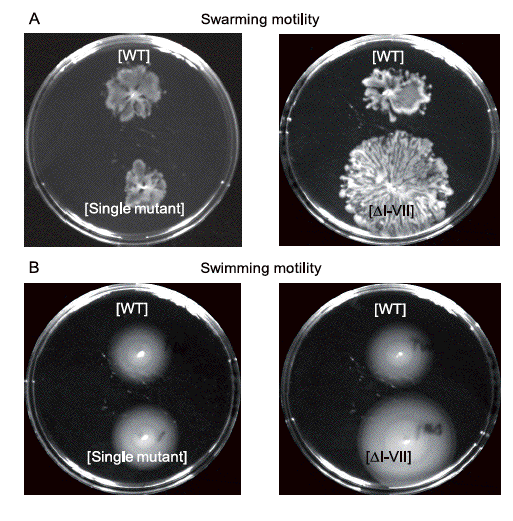
Effect of PAIs deletion on E. coli 536 motility (WT: wild type strain, DI-VII: mutant strain of all PAIs)
We have published a group of 3 papers showing the worrying potential of dissemination of extended spectrum beta-lactamase (ESBL)-producing E. coli strains in the hospital but also in the community, and eventually their virulence. In the july 2010 issue of the Journal of Clinical Microbiology, we investigated, in collaboration with our colleagues of Trousseau hospital, an extensive outbreak of a TEM-52-producing E. coli strain that colonized almost half of neonates in a ward, causing a meningitis in a newborn. In the july 2010 issue of the Journal of Antimicrobial Chemotherapy, we reported with our colleagues of Paul Brousse hospital an intrafamily transmission of a highly virulent CTX-M-3-producing E. coli clone causing an urinary tract infection in the mother and her adult son. Lastly, in the august 2010 issue of the Journal of Infectious Diseases, we showed in collaboration with the Antoine Andremont group that in a remote and controlled population of Amerindians living in the Trois-Sauts village located in the southernmost part of French Guiana, the ESBL-producing E. coli carriage prevalence was 8.0% in 2006, as compared to 3.2% in 2001.

ERIC-1 (a) and ERIC-2 (b) PCRs of CTX-M-3-producing E. coli isolates from urine and rectum of the mother (lines 1 and 2) and her son (lines 3 and 4)
In the march 2010 issue of Nature Reviews Microbiology, we have reviewed the population structure of commensal Escherichia coli and discussed how commensal strains can adapt to different niches and how commensalism can evolve into pathogenicity. Despite the occurrence of recombination events, the population structure is predominantly clonal, allowing the delineation of major phylogenetic groups. The genetic structure of commensal E. coli is shaped by multiple host and environmental factors, and the determinants involved in the virulence of the bacteria may in fact reflect adaptation to commensal habitats.
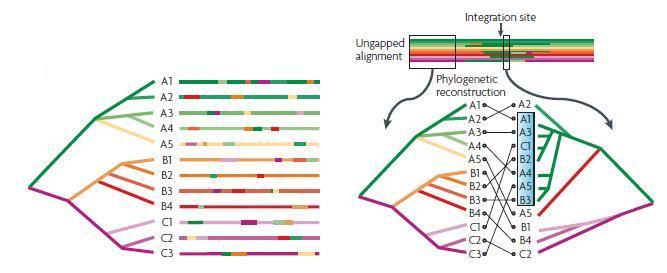
In the january 2009 issues of PLoS Genetics and PLoS Pathogens we showed in a multi-team collaborative work coordinated by our lab, using comparative genomics, phylogenetic and population genetics approaches that the E. coli genomes are subjected to a high level of gene flow, with a pan-genome (all the genes found in the strains) of almost 20,000 genes for a core genome (the genes present in all the strains) of less than 2,000 genes. We discussed the apparent paradox that, despite this high level of horizontal gene transfers, a robust species phylogeny can be inferred and genes co-exist in an organised genome. As a consequence, multiple evolutionary paths result in phenotypic diversification and there is no simple association between the presence of a gene and a given phenotype.
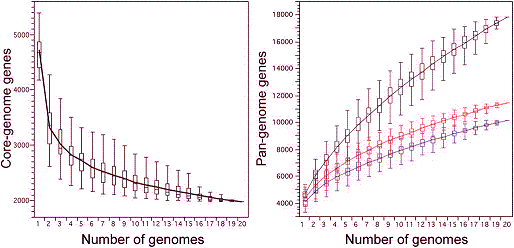
E. coli core and pan-genome evolution according to the number of sequenced genomes